µŹ«ń╗¤Ķ«Ī’╝īõ║║ń▒╗ĶøŗńÖĮõĖŁµ£ēµÄźĶ┐æ80%ĶøŗńÖĮµŚĀµśŠĶæŚµ┤╗µĆ¦õĮŹńé╣’╝īõ╝Āń╗¤õĖŖĶó½Ķ«żõĖ║µś»õĖŹÕÅ»µłÉĶŹ»ķØČńé╣ŃĆéõ╝Āń╗¤ńÜäķØȵĀćÕż¦ķā©Õłåµś»Õģʵ£ēµśÄńĪ«µ┤╗µĆ¦õĮŹńé╣ńÜäĶøŗńÖĮ’╝īõĖöķĆéÕÉłń╗ōÕÉłÕ░ÅÕłåÕŁÉ’╝īÕ╣ČõĖ╗Ķ”üķĆÜĶ┐ćÕŹĀµŹ«µ┤╗µĆ¦õĮŹńé╣ńÜäĶŹ»ńÉåÕŁ”õĮ£ńö©µ©ĪÕ╝Å (MOA) µØźµÄ¦ÕłČĶøŗńÖĮÕŖ¤ĶāĮŃĆéĶ┐Öń¦Źµ¢╣µ│ĢĶÖĮńäČń«ĆÕŹĢ’╝īõĮåµś»Õ╣ČõĖŹĶāĮÕ║öńö©õ║ĵēƵ£ēńö¤ńē®ķØȵĀć’╝īÕīģµŗ¼ķéŻõ║øń╝║õ╣Åķģȵ┤╗ŃĆüµøŠĶó½Ķ«żõĖ║µś»ŃĆīõĖŹÕÅ»µłÉĶŹ»ŃĆŹńÜäķØȵĀć’╝īõ╗źÕÅŖķĆÜĶ┐ćń¬üÕÅśÕÅŹÕżŹĶĆÉĶŹ»ŃĆüńÄ░ķśČµ«ĄµŚĀĶŹ»ÕÅ»µ▓╗ńÜäķØȵĀćŃĆéÕøĀµŁżķĢ┐µ£¤õ╗źµØź’╝īķØČÕÉæĶøŗńÖĮńÜäĶŹ»ńē®ńĀöÕÅæÕÅŚķÖÉõ║ĵ£ēķÖÉńÜäĶøŗńÖĮń¦Źń▒╗’╝īµ×üÕż¦Õ£░ķÖÉÕłČõ║åĶŹ»ńē®ÕÅæÕ▒ĢŃĆé
Ķć¬ĶĆČķ▓üÕż¦ÕŁ”ńÜäCraig CrewsµĢֵijÕøóķś¤ķ”¢µ¼ĪµŖźķüōõ║åÕ░ÅÕłåÕŁÉPROTACÕÉÄńÜäÕŹüÕ╣┤ķŚ┤’╝īPROTACķóåÕ¤¤ķŻ×ķƤÕÅæÕ▒ĢŃĆéĶŹ»ńē®ķØȵĀćńÜäµĀ╝Õ▒Ćõ╣¤ÕøĀµŁżÕÅæńö¤õ║åķćŹÕż¦ÕÅśÕī¢’╝īõ╗Äõ╝Āń╗¤ĶŹ»ńē®ķØȵĀćĶĮ¼ÕÉæµø┤ÕģʵīæµłśµĆ¦ńÜäķØȵĀćŃĆé
ń║ĄĶ¦éPROTACµŖƵ£»ÕÅæÕ▒ĢÕÄåÕÅ▓’╝ī2001Õ╣┤ķ”¢õĖ¬PROTACÕłåÕŁÉPROTAC-1Ķ»×ńö¤’╝īµś»ńö▒PROTACµŖƵ£»Õģłķ®▒CrewsÕÅŖÕģČÕÉīõ║ŗµŖźķüō’╝ø2008Õ╣┤’╝īń¼¼õĖĆõĖ¬Õ░ÅÕłåÕŁÉPROTACĶ»×ńö¤’╝īÕÉīµĀĘńö▒CrewsĶ»Šķóśń╗äĶ«ŠĶ«ĪÕÉłµłÉ’╝īµś»ńö▒ķØČÕÉæMDM2 E3µ│øń┤ĀĶ┐׵ğķģČńÜäÕ░ÅÕłåÕŁÉµŖæÕłČÕēénutlinÕÆīARÕ░ÅÕłåÕŁÉķģŹõĮōõ╗źÕÅŖõĖŁķŚ┤ńÜäPEG Linkerµ×䵳ɒ╝øõ╣ŗÕÉÄ’╝īńĀöń®ČĶĆģÕ╝ĆÕÅæÕć║õ╗źCRL4CRBNŃĆüCRL2VHLŃĆücIAPE3µ│øń┤ĀĶ┐׵ğķģČķģŹõĮōńÜäÕÉäń¦ŹÕ░ÅÕłåÕŁÉPROTACs’╝øÕåŹõ╣ŗÕÉÄ’╝īArvinasÕģ¼ÕÅĖńĀöÕÅæńÜäARV-110ÕÆīARV-471õĖżõĖ¬PROTACÕłåÕŁÉĶ┐øÕģźõĖ┤Õ║ŖÕ«×ķ¬ī’╝īÕłåÕł½ńö©õ║ĵ▓╗ń¢ŚÕēŹÕłŚĶģ║ńÖīÕÆīõ╣│Ķģ║ńÖīŃĆé
ÕøŠ’╝ÜPROTACķćīń©ŗńóæµŚČķŚ┤ńé╣
PROTAC µŖƵ£»ńÜäÕć║ńÄ░’╝īõ╗źÕģČķóĀĶ”åµĆ¦ńÜäĶ«ŠĶ«ĪŃĆüķóĀĶ”åµĆ¦ńÜäõĮ£ńö©µ£║ÕłČµīæµłśõ║åŃĆīõĖŹÕÅ»µłÉĶŹ»ŃĆŹķØČńé╣’╝īõĮ┐ĶĆÉĶŹ»µéŻĶĆģĶÄĘÕŠŚµ¢░õĖĆõ╗Żµ▓╗ń¢ŚĶŹ»ńē®ŃĆéĶ┐æõĖżÕ╣┤µØź’╝īPROTAC µŖƵ£»Ķ┐øÕģźķ½śķƤÕÅæÕ▒ĢķśČµ«Ą’╝īõĖ║ńö¤ńē®Õī╗ĶŹ»ńĀöÕÅæÕ╝Ƶŗōõ║åõĖĆńēćµ¢░ńÜäķóåÕ¤¤ŃĆé
õĖĆŃĆüµŖƵ£»ÕĤńÉåŌĆöŌĆöµ│øń┤Āõ╗ŗÕ»╝ńÜäĶøŗńÖĮķÖŹĶ¦Żń│╗ń╗¤
ĶøŗńÖĮĶ┤©µś»ń╗åĶā×ÕåģķćŹĶ”üńÜäńö¤ńē®Õż¦ÕłåÕŁÉ’╝īÕÅéõĖÄÕżÜń¦Źńö¤ÕæĮµ┤╗ÕŖ©’╝īÕīģµŗ¼ķģČÕÆīµ┐Ćń┤ĀńÜäÕŖ¤ĶāĮŃĆüĶ┐ÉÕŖ©ŃĆüĶ┐ÉĶŠōŃĆüÕģŹń¢½ÕÅŹÕ║öńŁēŃĆéõ╗źÕŠĆ’╝īõ║║õ╗¼ķØ×ÕĖĖÕģ│µ│©ĶøŗńÖĮĶ┤©Õ£©ń╗åĶā×Õåģµś»Õ”éõĮĢÕÉłµłÉńÜä’╝īõĮåÕ»╣õ║ÄĶøŗńÖĮĶ┤©Õ£©ń╗åĶā×Õåģµś»Õ”éõĮĢķÖŹĶ¦ŻńÜäÕŹ┤Õ░æµ£ēõ║║Õģ│µ│©ŃĆéõ╗źĶē▓ÕłŚń¦æÕŁ”Õ«ČAaron CiechanoverŃĆüAvram HershkoÕÆīńŠÄÕøĮń¦æÕŁ”Õ«ČIrwin RoseÕÅæńÄ░õ║åµ│øń┤Āõ╗ŗÕ»╝ńÜäĶøŗńÖĮĶ┤©ķÖŹĶ¦Żµ£║ÕłČ’╝īÕģ▒ÕÉīĶÄĘÕŠŚõ║å2004Õ╣┤Ķ»║Ķ┤ØÕ░öÕī¢ÕŁ”Õź¢ŃĆé
µ│øń┤Āµ£¼Ķ║½õ╣¤µś»õĖĆń¦ŹÕżÜĶéĮ’╝īńö▒76õĖ¬µ░©Õ¤║ķģĖµ«ŗÕ¤║ń╗䵳ɒ╝īÕ║ÅÕłŚķ½śÕ║”õ┐ØÕ«ł’╝īõĖöÕ£©ÕżÜń¦Źń£¤µĀĖńö¤ńē®õĖŁµÖ«ķüŹÕŁśÕ£©ŃĆéµ│øń┤ĀńÜäõĖ╗Ķ”üÕŖ¤ĶāĮµś»µĀćĶ«░ķ£ĆĶ”üÕłåĶ¦ŻńÜäĶøŗńÖĮĶ┤©’╝īÕĮōµ│øń┤ĀĶ┐×µÄźÕł░ĶøŗńÖĮõĖŖÕÉÄ’╝īõ╝ÜÕ»╝Ķć┤Ķ┐Öõ║øĶøŗńÖĮĶó½Ķ┐ÉķĆüÕł░ĶøŗńÖĮķģČõĮōõĖŁĶ┐øĶĪīķÖŹĶ¦Ż’╝īÕ«×ńÄ░ń╗åĶā×ÕåģĶøŗńÖĮńÜäÕ╣│ĶĪĪŃĆéµĆ╗ńÜäµØźĶ»┤’╝īµ│øń┤ĀÕÅéõĖÄõ║åń╗åĶā×Õ橵£¤ŃĆüÕó×µ«¢ŃĆüÕćŗõ║ĪŃĆüÕłåÕī¢ŃĆüĶĮ¼ń¦╗ŃĆüÕ¤║ÕøĀĶĪ©ĶŠŠŃĆüĶĮ¼ÕĮĢĶ░āĶŖéŃĆüõ┐ĪÕÅĘõ╝ĀķĆÆŃĆüµŹ¤õ╝żõ┐«ÕżŹŃĆüńéÄńŚćÕģŹń¢½ńŁēÕćĀõ╣ÄõĖĆÕłćńö¤ÕæĮµ┤╗ÕŖ©ńÜäĶ░āµÄ¦ŃĆé
µ│øń┤ĀÕī¢’╝īõĖ╗Ķ”üńö▒3ń¦ŹķģČÕé¼Õī¢’╝īÕŹ│E1µ│øń┤Āµ┤╗Õī¢ķģČŃĆüE2µ│øń┤Āń╗ōÕÉłķģČŃĆüE3Ķ┐׵ğķģČŃĆéÕ£©ATPńÜäõĮ£ńö©õĖŗ’╝īµ│øń┤Āõ╝ÜĶó½µ┐Ƶ┤╗’╝īÕģłÕĮóµłÉµ│øń┤Ā-Ķģ║ĶŗĘķģĖÕżŹÕÉłńē®’╝īÕåŹĶó½ĶĮ¼ń¦╗Õł░µ│øń┤Āµ┤╗Õī¢ķģČE1õĖŖ’╝øķÜÅÕÉÄ’╝īE1Õ░åµ┤╗Õī¢ńÜäµ│øń┤ĀĶĮ¼ń¦╗Õł░µ│øń┤Āń╗ōÕÉłķģČE2õĖŖ’╝øµ£Ćń╗ł’╝īµ│øń┤ĀĶ┐׵ğķģČE3Õ░åµ│øń┤ĀĶĮ¼ń¦╗Õł░ńø«µĀćĶøŗńÖĮõĖŖ’╝īÕ░▒Õāŵś»µēōõĖŖŌĆ£µĖģķÖżŌĆØńÜäµĀćńŁŠŃĆéĶ┐Öõ║øĶó½µēōõĖŖµĀćńŁŠńÜäńø«µĀćĶøŗńÖĮ’╝īõ╣¤õ╝ÜĶó½ķĆüÕł░ĶøŗńÖĮķģČõĮōÕżŹÕÉłõĮōÕżäĶ┐øĶĪīķÖŹĶ¦ŻŃĆé
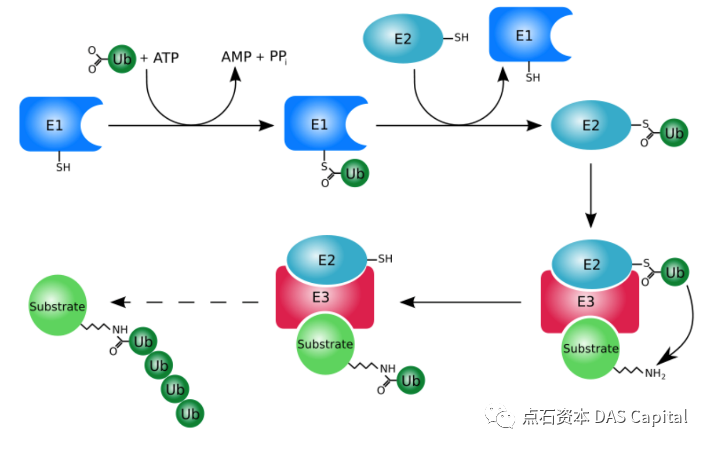
ÕøŠ’╝ܵ│øń┤ĀÕī¢ńż║µäÅÕøŠ
PROTAC’╝łPROteolysis TArgeting Chimeras’╝īĶøŗńÖĮķÖŹĶ¦ŻķØČÕÉæÕĄīÕÉłõĮō’╝ēńö▒õĖēń¦ŹÕģāń┤Āń╗䵳ɒ╝ÜE3µ│øń┤ĀĶ┐׵ğķģČķģŹõĮōŃĆüķØČĶøŗńÖĮķģŹõĮōÕÆīLinkerŃĆéE3µ│øń┤ĀĶ┐׵ğķģČķģŹõĮōĶ┤¤Ķ┤Żńē╣Õ╝éµĆ¦µŗøÕŗ¤E3µ│øń┤ĀĶ┐׵ğķģČ’╝øķØČĶøŗńÖĮķģŹõĮōńö©õ║ÄķØČÕÉæÕÆīµŹĢĶÄĘńø«µĀćĶøŗńÖĮ’╝øLinkerńö©õ║Äń╗ōÕÉłĶ┐ÖõĖżõĖ¬ķģŹõĮō’╝īÕĮóµłÉń©│Õ«ÜńÜäõĖēÕģāÕżŹÕÉłńē®ŃĆé
PROTACÕłåÕŁÉĶāĮÕż¤Õ░åE3µ│øń┤ĀĶ┐׵ğķģČÕŗ¤ķøåÕł░ķØČńé╣ĶøŗńÖĮķÖäĶ┐æ’╝īõĖ║ķØČńé╣ĶøŗńÖĮµēōõĖŖµ│øń┤ĀŌĆ£µĀćńŁŠŌĆØŃĆéÕ£©ń╗åĶā×õĖŁ’╝īµēōõĖŖµ│øń┤ĀŌĆ£µĀćńŁŠŌĆØńÜäĶøŗńÖĮÕ░åĶó½ķĆüÕģźĶøŗńÖĮķģČõĮōĶ┐øĶĪīķÖŹĶ¦ŻŃĆéķØČĶøŗńÖĮķÖŹĶ¦ŻÕÉÄ’╝īPROTACÕłåÕŁÉÕÅłÕÅ»õ╗źĶó½ķćŖµöŠÕć║µØźÕÅéõĖÄÕł░õĖŗõĖĆõĖ¬ĶøŗńÖĮńÜäķÖŹĶ¦ŻĶ┐ćń©ŗ’╝īÕøĀµŁżĶ┐Öń¦ŹķÖŹĶ¦ŻõĮ£ńö©Õģʵ£ēÕé¼Õī¢µĢłµ×£’╝īĶŠāÕ░æńÜäĶŹ»ńē®ÕēéķćÅÕ░▒ÕÅ»õ╗źÕ«×ńÄ░ķ½śµĢłńÜäķÖŹĶ¦ŻŃĆé
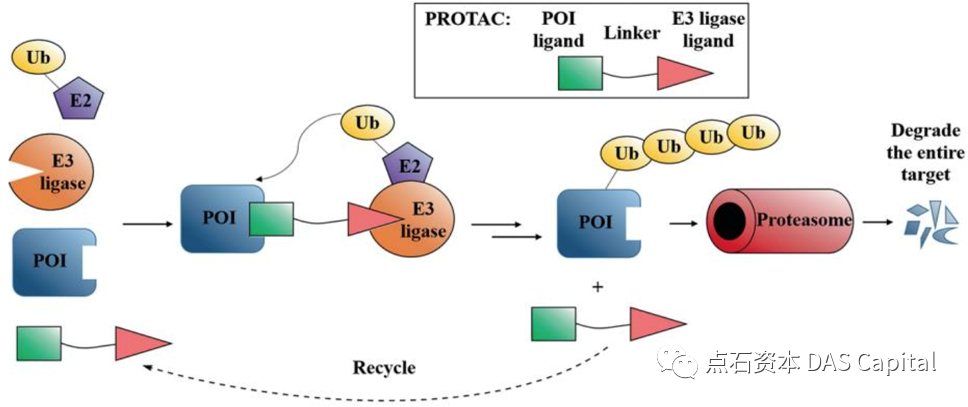
ÕøŠ’╝ÜPROTACķĆÜĶ┐ćµ│øń┤ĀĶ░āĶŖéńÜäĶøŗńÖĮķÖŹĶ¦ŻÕÅæµīźĶŹ»µĢł
ńø«ÕēŹ’╝īń╗åĶā×ÕåģķØČĶøŗńÖĮĶĪ©ĶŠŠµ░┤Õ╣│ńÜäĶ░āµÄ¦õĖ╗Ķ”üÕÅ»õ╗źõ╗ÄõĖēõĖ¬Õ▒éķØóĶ┐øĶĪīÕ╣▓ķóä:’╝ł1’╝ēÕ£©Õ¤║ÕøĀÕ▒éķØóĶ┐øĶĪīķØČĶøŗńÖĮńÜäĶ░āµÄ¦µŖƵ£»’╝īõŠŗÕ”éĶ┐æÕ╣┤Õģ┤ĶĄĘńÜäÕ¤║ÕøĀń╝¢ĶŠæµŖƵ£»(CRISPR-Cas9)’╝īõ╝śńé╣’╝Üń▓ŠńĪ«Õ║”ķ½śõĖöķĆÜńö©µĆ¦Õ╝║ńŁē’╝īń╝║ńé╣’╝ÜõĖŹĶāĮÕ»╣ķØČĶøŗńÖĮĶ┐øĶĪīÕŖ©µĆüĶ░āµÄ¦’╝īÕ╣ČÕģʵ£ēõĖŹÕÅ»ķĆåµĆ¦õ╗źÕÅŖÕŁśÕ£©µĮ£Õ£©ńÜäķüŚõ╝ĀĶĪźÕü┐µĢłÕ║ö;’╝ł2’╝ēÕ£©ĶĮ¼ÕĮĢÕ▒éķØóķĆÜĶ┐ćRNAÕ╣▓µē░µŖƵ£»ķØČÕÉæńē╣Õ«ÜńÜäķØČĶøŗńÖĮmRNA’╝īń╝║ńé╣’╝ܵĢłńÄćĶŠāõĮÄ’╝īõĖŹķĆéńö©õ║ÄńĀöń®ČĶŠāń©│Õ«ÜńÜäĶøŗńÖĮĶ┤©;
’╝ł3’╝ēÕ£©ĶøŗńÖĮÕ▒éķØóķĆÜĶ┐ćPROTAC µŖƵ£»Ķ┐øĶĪīķØČĶøŗńÖĮńÜäÕī¢ÕŁ”µĢ▓ķÖŹ’╝īĶ»źµ¢╣µ│ĢÕģʵ£ēķ½śµĢłŃĆüÕÅ»ķĆåńÜäõ╝śńé╣’╝īÕ╣ČõĖöĶāĮÕż¤Ķ┐øĶĪīÕé¼Õī¢ÕŠ¬ńÄ»’╝īÕÅ»õ╗źÕ£©õĮÄÕēéķćÅõĖŗÕÅæµīźõĮ£ńö©
õĖēŃĆüPROTACõ╝śÕŖ┐ÕÅŖÕ╝ĆÕÅæķÜŠńé╣
õĖÄõ╝Āń╗¤Õ░ÅÕłåÕŁÉµŗ«µŖŚÕēéńøĖµ»ö’╝īPROTACÕģʵ£ēõĖĆń│╗ÕłŚńŗ¼ńē╣ńÜäõ╝śÕŖ┐’╝Ü
’╝ł1’╝ēķĆēµŗ®µĆ¦µø┤ķ½śŃĆüõĮ£ńö©ķØČńé╣µø┤Õ╣┐
PROTACµ£ĆÕż¦ńÜäõ╝śÕŖ┐õ╣ŗõĖƵś»ĶāĮÕż¤õĮ┐ķØČńé╣õ╗ÄŌĆ£õĖŹÕÅ»µłÉĶŹ»µĆ¦ŌĆØÕÅśµłÉŌĆ£ÕÅ»µłÉĶŹ»µĆ¦ŌĆØŃĆéÕżÜµĢ░Õ░ÅÕłåÕŁÉĶŹ»ńē®µł¢ÕŹĢµŖŚķ£ĆĶ”üń╗ōÕÉłķģȵł¢ÕÅŚõĮōńÜäµ┤╗µĆ¦õĮŹńé╣µØźÕÅæµīźõĮ£ńö©’╝īõĮåµŹ«õ╝░Ķ«Ī’╝īõ║║ń▒╗ń╗åĶā×õĖŁ80%ńÜäĶøŗńÖĮń╝║õ╣ÅĶ┐ÖµĀĘńÜäõĮŹńé╣ŃĆéPROTACµŚĀķ£ĆõĖÄńø«µĀćĶøŗńÖĮķĢ┐µŚČķŚ┤ÕÆīķ½śÕ╝║Õ║”ńÜäń╗ōÕÉł’╝īõŠ┐ÕÅ»µŹĢĶÄĘĶøŗńÖĮÕ╣ČÕ░åÕģČķÖŹĶ¦Ż’╝īÕøĀµŁżµ£ēµ£øń¬üńĀ┤õ╝Āń╗¤ķÜŠõ╗źµłÉĶŹ»ńÜäķØČńé╣Õ╣ČÕģŗµ£ŹĶĆÉĶŹ»µĆ¦ķŚ«ķóśŃĆé
’╝ł2’╝ēÕé¼Õī¢ķÖŹĶ¦ŻÕŖ¤ĶāĮŃĆüµø┤õĮÄńÜäĶŹ»ńē®ÕēéķćÅ
õ╝Āń╗¤ńÜäÕ░ÅÕłåÕŁÉķććńö©ńÜäĶŹ»ńÉåÕŁ”õĮ£ńö©µ©ĪÕ╝ÅõĖ║ÕŹĀµŹ«ķ®▒ÕŖ©µ©ĪÕ╝Å’╝īõĖ║õ║åµÅÉķ½śķØČńé╣ÕŹĀµ£ēńÄć’╝īÕŠĆÕŠĆķ£ĆĶ”üķ½śÕēéķćÅńÜäĶŹ»ńē®’╝īÕÅ»ĶāĮõ╝ÜÕĖ”µØźĶŠāÕż¦ńÜäµ»ÆÕē»õĮ£ńö©ŃĆéPROTACķććńö©ńÜ䵜»õ║ŗõ╗Čķ®▒ÕŖ©’╝łevent-driven’╝ēńÜäĶŹ»ńÉåÕŁ”õĮ£ńö©µ©ĪÕ╝Å’╝īÕģČÕ»╣ńø«ńÜäĶøŗńÖĮńÜäķÖŹĶ¦ŻĶ┐ćń©ŗµś»õĖĆń¦ŹÕé¼Õī¢õĮ£ńö©’╝īÕøĀµŁżÕŬķ£ĆĶŠāõĮÄńÜäÕī¢ÕÉłńē®µĄōÕ║”õŠ┐ÕÅ»õ╗źĶŠŠÕł░ÕŠłÕźĮńÜäķÖŹĶ¦ŻµĢłńÄć’╝īõĖöĶāĮÕż¤ķÖŹĶ¦ŻµĢ┤õĖ¬ĶøŗńÖĮŃĆé
’╝ł3’╝ēÕ╗ČķĢ┐õĮ£ńö©µŚČķŚ┤
ķØČĶøŗńÖĮńÜäķÖŹĶ¦Żµś»µŚČķŚ┤õŠØĶĄ¢µĆ¦ńÜä’╝īPROTACÕÅ»õ╗źÕ£©ÕćĀÕłåķƤÕåģÕ░åĶā×ÕåģķØČĶøŗńÖĮµČłĶĆŚÕł░µÄźĶ┐æÕ¤║ńĪƵ░┤Õ╣│ŃĆéÕĮōÕĤÕģłÕŁśÕ£©ńÜäķØČĶøŗńÖĮĶĆŚÕ░ĮÕÉÄ’╝īPROTACÕŬķ£ĆķÖŹĶ¦Żķ揵¢░ÕÉłµłÉńÜäķØČĶøŗńÖĮ’╝īÕż¦ÕżÜµĢ░ĶøŗńÖĮĶ┤©ńÜäÕåŹÕÉłµłÉķƤÕ║”ÕŠłµģó’╝īÕŹ│õĮ┐Õ£©PROTACÕ«īÕģ©µĖģķÖżÕÉÄ’╝īń╗åĶā×ÕÅ»ĶāĮõ╗Źķ£ĆĶ”üõĖƵ«ĄńøĖÕĮōķĢ┐ńÜ䵌ČķŚ┤’╝īµēŹĶāĮÕ░åĶøŗńÖĮĶ┤©µüóÕżŹµŁŻÕĖĖńö¤ńÉåµ░┤Õ╣│’╝īõ╗ÄĶĆīÕż¦Õż¦Õ╗ČķĢ┐õĮ£ńö©µŚČķŚ┤ŃĆé

ĶĪ©’╝ÜPROTACõĖÄÕģČõ╗¢ĶŹ»ńē®Õ»╣µ»ö
õĮåPROTACÕ╝ĆÕÅæķÜŠÕ║”ĶŠāÕż¦’╝īńø«ÕēŹÕ£©ńĀöńÜäPROTACÕŁśÕ£©ÕÅŻµ£ŹÕÉĖµöČÕÅŖķĆÅĶå£µĆ¦ĶŠāÕĘ«ńŁēķŚ«ķóśŃĆéõĖƵ¢╣ķØó’╝īPROTACĶŹ»ńē®Õ▒×õ║ÄÕÅīķØČńé╣ĶŹ»ńē®’╝īÕłåÕŁÉķćÅĶŠāÕż¦’╝īń╗ōµ×äÕżŹµØé’╝īĶŹ»õ╗ŻÕŖ©ÕŖøÕŁ”Õ╣ČõĖŹõ╣ÉĶ¦é’╝īÕÅŻµ£ŹÕÉĖµöČÕÆīķĆÅĶå£µĆ¦ÕŁśÕ£©µīæµłś’╝øÕÅ”õĖƵ¢╣ķØó’╝īPROTACķØȵĀćĶøŗńÖĮķÖŹĶ¦ŻĶāĮÕŖøµśŠĶæŚ’╝īõĮåńö▒õ║ÄķØČĶøŗńÖĮÕ£©µŁŻÕĖĖń╗åĶā×õĖŁõ╣¤õ╝ÜĶĪ©ĶŠŠ’╝īPROTACÕ»╣µŁŻÕĖĖń╗äń╗ćńÜäµ»ÆµĆ¦ķĪ╗Õ»åÕłćÕģ│µ│©ŃĆ鵣żÕż¢’╝īńö©õ║ÄÕ░ÅÕłåÕŁÉĶŹ»ńē®µŚ®µ£¤ńŁøķĆēńÜäLipinskińÜäŌĆ£õ║öµ│ĢÕłÖŌĆØÕ╣ČõĖŹķĆéńö©õ║ÄPROTACķóåÕ¤¤ŃĆé
ńø«ÕēŹPROTACķØČńé╣õĖ╗Ķ”üÕłåõĖ║Õ”éRASŃĆüSHP2ńŁēķÜŠµłÉĶŹ»ń▒╗ńÜäķØČńé╣’╝īõ╗źÕÅŖBTKŃĆüEGFRķØČÕÉæµ▓╗ń¢ŚÕÉÄÕżŹÕÅæĶĆÉĶŹ»ńÜäķØČńé╣ŃĆé
a)┬Ā┬Ā┬Ā┬Ā┬ĀķÜŠµłÉĶŹ»ķØČńé╣
┬Āi.┬Ā ┬Ā┬ĀRAS
RASµś»ńÖīńŚćõĖŁµ£ĆÕĖĖĶ¦üńÜäń¬üÕÅśÕ¤║ÕøĀ’╝īÕ£©Ķé║ńÖīŃĆüń╗ōńø┤ĶéĀńÖīÕÆīĶā░Ķģ║ńÖīõĖŁÕØćÕŁśÕ£©Ķ┐ćĶĪ©ĶŠŠµł¢µ┤╗Õī¢ŃĆéÕ£©Ķó½ÕÅæńÄ░ÕÆīńĀöń®ČńÜä40ÕżÜÕ╣┤µØź’╝īõĖĆńø┤µ▓Īµ£ēķÆłÕ»╣Ķ»źķØČńé╣ńÜäĶŹ»ńē®õĖŖÕĖé’╝īķĢ┐µ£¤Ķó½ń¦░õĖ║ŌĆ£õĖŹÕÅ»µłÉĶŹ»ŌĆØńÜäķØČńé╣ŃĆéĶÖĮńäČÕÄ╗Õ╣┤5µ£łõ╗ĮõĖŖÕĖéńÜäAMG510’╝łSotorasib’╝ēń╗łń╗ōõ║åĶ»źķØČńé╣ńÜäŌĆ£õĖŹÕÅ»µłÉĶŹ»µĆ¦ŌĆØ’╝īõĮåµś»ķÖŹĶ¦ŻÕēéÕ£©Ķ»źķØČńé╣õ╗ŹńäČÕż¦µ£ēÕÅ»õĖ║ŃĆé
ķÆłÕ»╣KRAS-G12Cń¬üÕÅś’╝ī2020Õ╣┤PROTACsµŖƵ£»Õģłķ®▒Craig CrewsµĢֵijÕĖ”ķóåńÜäńĀöń®ČÕ░Åń╗äķ”¢µ¼ĪµŖźķüōõ║åfirst-in-classńÜäÕåģµ║ɵƦKRAS-G12CķÖŹĶ¦ŻÕēéLC-2ńÜäÕ╝ĆÕÅæŃĆéńĀöń®ČÕÅæńÄ░’╝īLC-2ÕÅ»Õ┐½ķƤŃĆüµīüń╗ŁÕ£░ķÖŹĶ¦Żń║»ÕÉłÕÆīµØéÕÉłń¬üÕÅśń╗åĶā×ń│╗õĖŁńÜäKRAS-G12C’╝īÕ»╝Ķć┤MAPKõ┐ĪÕÅĘõ╝ĀÕ»╝ÕÅŚÕł░µŖæÕłČ’╝īĶĪ©µśÄPROTACsõ╗ŗÕ»╝ńÜäķÖŹĶ¦Żµś»ĶĪ░ÕćÅńÖīń╗åĶā×õĖŁĶć┤ńÖīKRASµ░┤Õ╣│ÕÆīõĖŗµĖĖõ┐ĪÕÅĘńÜäõĖĆń¦ŹÕÅ»ĶĪīńŁ¢ńĢźŃĆé
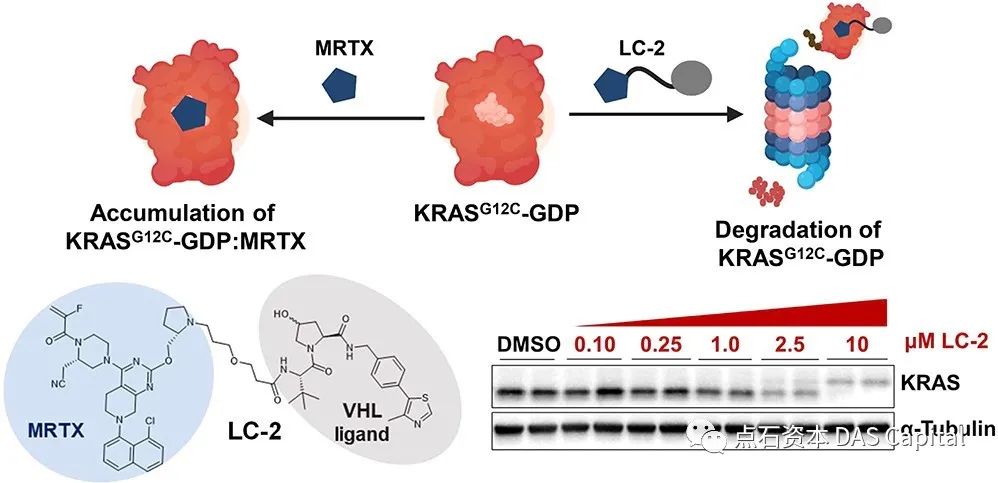
ÕøŠ’╝Üķ”¢õĖ¬µłÉÕŖ¤õ╗ŗÕ»╝Õåģµ║ÉKRAS G12CķÖŹĶ¦ŻńÜäPROTACÕłåÕŁÉLC-2
Source: ACSCent. Sci. 2020, 6, 8, 1367-1375
ÕÅ»õ╗źķóäĶ¦ü’╝īÕł®ńö©PROTACµŖƵ£»µö╗ÕģŗńÜäÕ░åõĖŹÕŬµś»G12C’╝īÕ»╣G12DńŁēÕģČõ╗¢ń¬üÕÅśõ╣¤Õ░åÕż¦µ£ēÕÅ»õĖ║ŃĆé
┬Āb)┬ĀĶŹ»ńē®Õ»╝Ķć┤ńÜäń¬üÕÅśĶĆÉĶŹ»
Õ░ÅÕłåÕŁÉµŖæÕłČÕē鵳¢µŗ«µŖŚÕēéÕ£©õĖ┤Õ║Ŗńö©ĶŹ»Ķ┐ćń©ŗõĖŁ’╝īõĖŹÕÅ»ķü┐ÕģŹńÜäķāĮõ╝ÜÕÅæńö¤ĶÄĘÕŠŚµĆ¦ĶĆÉĶŹ»ŃĆéµ»öÕ”éEGFR-T790MÕÆīC797SĶĆÉĶŹ»ńŁēŃĆéĶÖĮńäČÕÅ»õ╗źķĆÜĶ┐ćÕ╝ĆÕÅæµ¢░õĖĆõ╗ŻńÜäµŖæÕłČÕēéĶ¦ŻÕå│ĶĆÉĶŹ»ķŚ«ķóś’╝īõĮåµś»ķÜÅńØƵ¢░õĖĆõ╗ŻĶŹ»ńē®ńÜäõĮ┐ńö©’╝īµ¢░ńÜäĶĆÉĶŹ»õ╣¤õ╝ÜķÜÅõ╣ŗÕć║ńÄ░ŃĆé
i.┬Ā┬Ā┬Ā┬Ā EGFR
EGFR’╝łEpidermal Growth Factor Receptor’╝ēµś»õĖŖńÜ«ńö¤ķĢ┐ÕøĀÕŁÉ’╝łEGF’╝ēń╗åĶā×Õó×µ«¢ÕÆīõ┐ĪÕÅĘõ╝ĀÕ»╝ńÜäÕÅŚõĮō’╝īÕł║µ┐ĆõĖŗµĖĖńÜäPI3K/AKTķĆÜĶĘ»ŃĆüRAS/RAFķĆÜĶĘ»’╝īÕ»╣ń╗åĶā×ńÜäńö¤ķĢ┐ŃĆüÕó×µ«¢ŃĆüÕłåÕī¢õ║¦ńö¤ķćŹĶ”üõĮ£ńö©ŃĆé
EGFRńÜäĶ┐ćÕ║”ĶĪ©ĶŠŠÕÆīń¬üÕÅśõĖÄķØ×Õ░Åń╗åĶā×Ķé║ńÖī’╝łNSCLC’╝ēńÜäÕÅæńö¤ÕÅæÕ▒ĢÕ»åÕłćńøĖÕģ│’╝īõ╣¤µś»NSCLCķćŹĶ”üńÜäĶŹ»ńē®ķØČńé╣õ╣ŗõĖĆŃĆéńø«ÕēŹÕøĮÕåģÕĘ▓õĖŖÕĖéõĖēõ╗ŻEGFRķØČÕÉæµŖæÕłČÕēé’╝īÕīģµŗ¼’╝ł1’╝ēń¼¼õĖĆõ╗ŻEGFRµŖæÕłČÕēé’╝ÜķØČÕÉæ19-Del/21-L858Rń¬üÕÅśńÜäÕÄäµ┤øµø┐Õ░╝ŃĆüÕÉēķØ×µø┐Õ░╝ŃĆüÕ¤āÕģŗµø┐Õ░╝’╝ī’╝ł2’╝ēń¼¼õ║īõ╗ŻEGFRµŖæÕłČÕēé’╝ÜõĖÄÕ║Ģńē®õĖŹÕÅ»ķĆåń╗ōÕÉłńÜäķś┐µ│Ģµø┐Õ░╝ŃĆüĶŠŠÕģŗµø┐Õ░╝’╝ī’╝ł3’╝ēń¼¼õĖēõ╗ŻEGFRµŖæÕłČÕēé’╝ÜÕ£©ķØČÕÉæ19-Del/21-L858Rń¬üÕÅśńÜäÕ¤║ńĪĆõĖŖÕÉīµŚČķØČÕÉæ20-T790Mń¬üÕÅśńÜäÕźźÕĖīµø┐Õ░╝ÕÆīķś┐ńŠÄµø┐Õ░╝ŃĆéõĮå15%~32%ńÜäµéŻĶĆģõ╝ÜÕÅæńö¤ń¼¼õĖēµ¼ĪEGFRń¬üÕÅś’╝īÕ╣ȵ£ĆÕĖĖÕÅæńö¤Õ£©EGFR C797SõĮŹńé╣’╝īÕŹ│ÕźźÕĖīµø┐Õ░╝õĖÄEGFRńÜäń╗ōÕÉłõĮŹńé╣ŃĆé
ķØóÕ»╣C797SõĮŹńé╣ń¬üÕÅś’╝īńø«ÕēŹõĖ┤Õ║ŖõĖŖÕ░ÜÕżäõ║ĵŚĀĶŹ»ÕÅ»µ▓╗ńŖȵĆüŃĆéĶĆīÕł®ńö©PROTACµŖƵ£»Õ╝ĆÕÅæńÜäEGFRķØČÕÉæĶøŗńÖĮķÖŹĶ¦ŻÕłåÕŁÉµ£ēµ£øÕĖ”µØźÕĘ©Õż¦õĖ┤Õ║ŖĶÄĘńøŖŃĆé
ńø«ÕēŹ’╝īµłæõ╗¼µ»ÅÕ╣┤µ¢░ÕÅæķØ×Õ░Åń╗åĶā×Ķé║ńÖīµéŻĶĆģõ║║µĢ░ĶČģĶ┐ć60õĖćõ║║’╝īÕģČõĖŁµÉ║ÕĖ”EGFRń¬üÕÅśńÜäµéŻĶĆģÕŹĀµ»öķ½śĶŠŠ40%ŃĆéµĀ╣µŹ«Õ╝ŚĶŗźµ¢»ńē╣µ▓ÖÕł®µ¢ćµĢ░µŹ«’╝ī2019Õ╣┤õĖŁÕøĮEGFR-TKI ĶŹ»ńē®ÕĖéÕ£║ÕĘ▓ń╗ÅµÄźĶ┐æ80õ║┐Õģāõ║║µ░æÕĖü’╝īķóäĶ«Ī2022Õ╣┤õĖŁÕøĮEGFR-TKI ĶŹ»ńē®ÕĖéÕ£║Ķ¦äµ©ĪÕ░åĶČģĶ┐ć200õ║┐’╝īCAGRõ┐صīü30%õ╗źõĖŖŃĆé
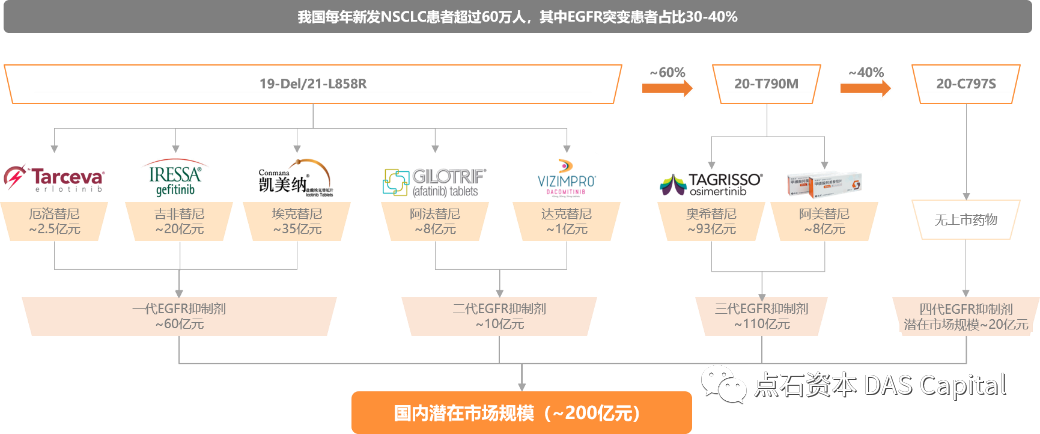
ĶĪ©’╝ÜķØ×Õ░Åń╗åĶā×Ķé║ńÖīEGFRÕĖéÕ£║Ķ¦äµ©ĪµĄŗń«Ś
µĢ░µŹ«µØźµ║É’╝Üńé╣ń¤│µĢ┤ńÉå
Craig M. CrewsŃĆüķćæÕüźŃĆüÕ╝ĀõĖēÕźćŃĆüõĖüÕģŗńŁēĶ»Šķóśń╗äķāĮµ£ēńøĖÕģ│EGFR-PROTACsńÜäńĀöń®Č’╝īńĀöń®Čõ║║ÕæśÕ░åń¼¼õĖĆŃĆüõ║īŃĆüõĖēõ╗ŻµŖæÕłČÕēéõĮ£õĖ║ń╗ōÕÉłķØČĶøŗńÖĮEGFRńÜäķģŹõĮōÕ║öńö©Õł░PROTACńÜäĶ«ŠĶ«ĪõĖŁ’╝īµŚ©Õ£©ķĆÜĶ┐ćĶøŗńÖĮķÖŹĶ¦ŻķĆöÕŠäÕģŗµ£ŹĶĆÉĶŹ»ń¬üÕÅśµł¢µēŠÕ»╗ń¬üńĀ┤µŖæÕłČÕēéńÜäĶøŗńÖĮķÖŹĶ¦Żń¢Śµ│ĢŃĆéPROTACń¢Śµ│ĢńÜäÕ║öńö©’╝īµ£ēµ£øĶ¦ŻÕå│ńö©ĶŹ»ÕÉÄEGFRõ║¦ńö¤µ¼Īń║¦ń¬üÕÅśńÜäķŚ«ķóś’╝īÕćÅÕ░æĶĆÉĶŹ»ÕÅæńö¤’╝īÕÉīµŚČĶāĮÕż¤ķ½śµĢłķØČÕÉæÕżÜń¦ŹEGFRń¬üÕÅś’╝īµø┐õ╗ŻõĖĆŃĆüõ║īŃĆüõĖēõ╗ŻEGFRõ╝Āń╗¤Õ░ÅÕłåÕŁÉµŖæÕłČÕēé’╝īµÄ©ĶĪīĶć│õĖĆŃĆüõ║īń║┐µ▓╗ń¢Śµ¢╣µĪłŃĆé
ķÆłÕ»╣EGFRÕżÜń¬üÕÅśPROTACĶŹ»ńē®ńÜäńĀöÕÅæÕ╣ČõĖŹÕ«╣µśō’╝īńö▒õ║ÄõĮŹńé╣ķĆēµŗ®µ£ēķÖÉŃĆüÕ»╣ķĆēµŗ®µĆ¦õĖÄÕ«ēÕģ©µĆ¦ńÜäÕÅ¢ĶłŹŃĆüÕÉłµłÉÕĘźĶē║ÕżŹµØéńŁēÕżÜµ¢╣ķØóÕøĀń┤ĀÕłČń║”’╝īńø«ÕēŹÕģ©ńÉāÕ░ܵŚĀõ║¦ÕōüĶ┐øÕģźõĖ┤Õ║ŖķśČµ«ĄŃĆéC4 TherapeuticsµŁŻÕ£©Õ╝ĆÕÅæńÜäÕĆÖķĆēĶŹ»ńē®CFT8919ķØČÕÉæEGFR L858Rń¬üÕÅśńÜäÕÅśµ×äõĮŹńé╣’╝īõĖ┤Õ║ŖÕēŹµĢ░µŹ«µśŠńż║ÕģČĶāĮµśŠĶæŚÕćÅÕ░æEGFRµ¼Īń║¦ń¬üÕÅś’╝īõ╗ÄĶĆīÕćÅÕ░æĶĆÉĶŹ»ÕÅæńö¤’╝īĶ»źĶŹ»ńē®Õ»╣C797SÕÆīT790Mń¬üÕÅśÕÉīµĀĘÕģʵ£ēµ┤╗µĆ¦’╝īÕ╣ČÕģʵ£ēÕģźĶäæĶāĮÕŖø’╝īµ£ēµ£øÕ»╣ĶäæĶĮ¼ń¦╗Ķé┐ńśżµ£ēµĢłŃĆéÕģ¼ÕÅĖĶ«ĪÕłÆ2022Õ╣┤õĖŁńö│µŖźIND’╝ī2022Õ╣┤Õ║ĢÕ╝ĆÕÉ»õĖ┤Õ║ŖĶ»Ģķ¬īŃĆé
ii.┬Ā ┬Ā┬Ā┬ĀBTK
BTKµ┐Ćķģȵś»BCRõ┐ĪÕÅĘķĆÜĶĘ»ńÜäÕģ│ķö«ĶøŗńÖĮ’╝īÕ»╣Bń╗åĶā×ńÜäÕó×µ«¢ŃĆüÕŁśµ┤╗Õģʵ£ēķćŹĶ”üõĮ£ńö©ŃĆéBTKĶøŗńÖĮń╝║Õż▒õ╝ÜÕ»╝Ķć┤µłÉńå¤Bń╗åĶā×µŚĀµ│Ģõ║¦ńö¤ŃĆéõ╝ŖÕĖāµø┐Õ░╝õĮ£õĖ║ķ”¢õĖ¬õĖŖÕĖéńÜäBTKµŖæÕłČÕēé’╝īõ║Ä2013Õ╣┤Õ£©ńŠÄÕøĮõĖŖÕĖéÕÉÄ’╝īõĖĆÕ╣┤ÕåģõŠ┐µłÉõĖ║ķöĆÕö«ķóØÕó×ķĢ┐µ£ĆÕ┐½ńÜäµŖŚĶé┐ńśżĶŹ»ńē®õ╣ŗõĖĆŃĆéõĮåµś»’╝īIbrutinibõ╣¤ÕŁśÕ£©õĖżõĖ¬µśÄµśŠńÜäÕ╝▒ńé╣’╝ÜõĖƵ¢╣ķØ󵜻off-targetÕ»╝Ķć┤ńÜäÕē»õĮ£ńö©’╝øÕÅ”õĖƵ¢╣ķØ󵜻BTKńÜäń¬üÕÅśõĮōC481SÕ»╝Ķć┤ńÜäĶĆÉĶŹ»ŃĆé
BTKõ╗ÄõĖĆõ╗ŻµŖæÕłČÕēéÕł░ńÄ░Õ£©ńÜäõ║īõ╗ŻµŖæÕłČÕēé’╝īĶÖĮńäČĶŹ»µĢłķāĮķØ×ÕĖĖµśÄµśŠ’╝īõĮåµ×üµśōõ║¦ńö¤ĶĆÉĶŹ»ŃĆéõ╗źõĖĆõ╗ŻBTKµŖæÕłČÕēéõĖ║õŠŗ’╝īµ»Å24Õ░ŵŚČõŠ┐µ£ēńŚģõ║║ńÜäBTKĶøŗńÖĮµ░┤Õ╣│µüóÕżŹÕł░µ▓╗ń¢ŚÕēŹńÜä3%Ķć│30%õ╗źõĖŖŃĆé
ÕøĀµŁż’╝īõĖ┤Õ║ŖµĆźķ£ĆĶāĮÕż¤Õģŗµ£ŹBTKń¬üÕÅśĶĆÉĶŹ»õĖöÕģʵ£ēµø┤Õ░ÅÕē»õĮ£ńö©ńÜäĶŹ»ńē®ŃĆéķććńö©PROTACµŖƵ£»’╝īõĖƵś»ÕøĀõĖ║Õ«āńÉåĶ«║õĖŖÕÅ»õ╗źÕŠ¬ńÄ»Õł®ńö©’╝īÕĮōPROTACĶ┐øÕģźń╗åĶā×ÕÉÄ’╝īÕÅ»õ╗źµīüń╗ŁÕ£░ķÖŹĶ¦Żµ¢░ńö¤µł¢ÕĤµ£ēńÜäBTKĶøŗńÖĮ’╝øń¼¼õ║ī’╝īÕŬĶ”üõŠØĶĄ¢BTK’╝īµŚĀĶ«║µś»µĆĵĀĘńÜäń╗ōÕÉłµ¢╣Õ╝Å’╝īķÖŹĶ¦Żõ╣ŗÕÉÄ’╝īÕģČõ╗¢ńÜäõĮ£ńö©µ£║ÕłČķāĮµŚĀµ│ĢĶĄĘõĮ£ńö©ŃĆé
õ║öŃĆüÕøĮÕåģÕż¢ńøĖÕģ│õ╝üõĖÜ
’╝łõĖĆ’╝ē┬Ā┬Ā┬Ā µĄĘÕż¢

Arvinasńö▒CrewsÕ£©2013Õ╣┤Õłøń½ŗ’╝īµś»µ£ĆµŚ®ÕĖāÕ▒ĆPROTACńÜäÕģ¼ÕÅĖõ╣ŗõĖĆ’╝īÕ╝ĆÕÅæńÜäĶøŗńÖĮķÖŹĶ¦ŻµŖƵ£»õĖ╗Ķ”üńö©õ║ÄĶé┐ńśżÕÆīńź×ń╗Åń│╗ń╗¤ń▒╗ń¢ŠńŚģńÜäµ▓╗ń¢ŚŃĆéńø«ÕēŹ’╝īÕģ¼ÕÅĖÕĘ▓µ£ēõĖżµ¼ŠPROTACĶŹ»ńē®Ķ┐øÕģźõĖ┤Õ║ŖIIµ£¤’╝īÕłåÕł½µś»ķØČÕÉæķøäµ┐Ćń┤ĀÕÅŚõĮō’╝łAR’╝ēńÜäARV-110ÕÆīķØČÕÉæķøīµ┐Ćń┤ĀÕÅŚõĮō’╝łER’╝ēńÜäARV-471ŃĆé
2021Õ╣┤7µ£łĶŠēńæ×õĖÄArvinasĶŠŠµłÉÕŹÅĶ««’╝īÕģ▒ÕÉīÕ╝ĆÕÅæÕ╣ČÕĢåõĖÜÕī¢ARV-471ŃĆéµĀ╣µŹ«ÕŹÅĶ««’╝īArvinasÕ░åĶÄĘÕŠŚ6.5õ║┐ńŠÄÕģāńÜäķóäõ╗śµ¼Š’╝īõ╗źÕÅŖÕżÜĶŠŠ14õ║┐ńŠÄÕģāńÜäķćīń©ŗńóæõ╗śµ¼Š’╝īĶŠēńæ×Ķ┐śÕ░åÕ»╣ArvinasĶ┐øĶĪī3.5õ║┐ńŠÄÕģāńÜäĶéĪµØāµŖĢĶĄäŃĆéķÖżĶŠēńæ×Õż¢’╝īArvinasĶ┐śõĖÄķ╗śµ▓ÖõĖ£ŃĆüÕ¤║ÕøĀµ│░ÕģŗŃĆüµŗ£ĶĆ│ńŁēÕłČĶŹ»ÕĘ©Õż┤Õ╗║ń½ŗõ║åÕÉłõĮ£Õģ│ń│╗ŃĆé
C4 Therapeuticsńö▒James BradnerµłÉń½ŗõ║Ä2015Õ╣┤’╝īĶ»źÕģ¼ÕÅĖµŗźµ£ēõĖōµ│©õ║ÄĶøŗńÖĮķÖŹĶ¦ŻÕēéÕ╝ĆÕÅæńÜäÕ╣│ÕÅ░C4T TORPEDO’╝īńö©õ║ÄPROTACńÜäĶ«ŠĶ«ĪŃĆüÕÉłµłÉÕÆīµ┤╗µĆ¦Ķ»äõ╗Ę’╝īµŚ©Õ£©ÕÅæńÄ░ķ½śĶ┤©ķćÅńÜäĶøŗńÖĮķÖŹĶ¦ŻÕēéŃĆé
ńø«ÕēŹÕĖāÕ▒ĆńÜäķØČńé╣õĖÄĶé┐ńśżńøĖÕģ│’╝īÕ”éIKZF1/3ŃĆüBRD9ŃĆüEGFRŃĆüBRAF-V600EÕÆīRETńŁēŃĆé2019Õ╣┤1µ£ł’╝īC4 TherapeuticsõĖĵĖżÕüźÕÆīńĮŚµ░ÅÕłåÕł½ĶŠŠµłÉ4.15õ║┐ńŠÄÕģāÕÆī9õ║┐ńŠÄÕģāńÜäÕÉłõĮ£ÕŹÅĶ««ŃĆé
Kymera TherapeuticsµłÉń½ŗõ║Ä2016Õ╣┤’╝īõĖōµ│©õ║Äńö©ĶøŗńÖĮķÖŹĶ¦ŻµŖƵ£»µ▓╗ń¢ŚńÖīńŚćÕÆīÕģŹń¢½µĆ¦ńéÄńŚć’╝īÕ╗║ń½ŗõ║åńŗ¼ńē╣ńÜäPegasusÕ╣│ÕÅ░’╝īĶ»źÕ╣│ÕÅ░ÕÅ»Õł®ńö©ńö¤ńē®õ┐Īµü»ÕŁ”ķ®▒ÕŖ©ķØȵĀćńÜäÕÅæńÄ░ŃĆüÕ╗║ń½ŗńÜäE3Ķ┐׵ğķģČÕ║ōŃĆüµŗźµ£ēÕģłĶ┐øńÜäĶøŗńÖĮķÖŹĶ¦ŻµŻĆµĄŗµēŗµ«ĄÕÆīķ½śµ░┤ÕćåńÜäń╗ōµ×äńö¤ńē®ÕŁ”µ©ĪÕ×ŗŃĆéÕĖāÕ▒ĆńÜäķØČńé╣µ£ēIRAK4ŃĆüSTAT3ńŁē’╝īĶ┐øÕ▒Ģµ£ĆÕ┐½ńÜ䵜»KT-474’╝īńø«ÕēŹÕ£©Iµ£¤õĖ┤Õ║ŖŃĆé
Ķ»źÕģ¼ÕÅĖńÜäÕÉłõĮ£õ╝Öõ╝┤õĖ╗Ķ”üµś»ĶĄøĶ»║ĶÅ▓ÕÆīĶæøÕģ░ń┤ĀÕÅ▓ÕģŗńŁēŃĆé2020Õ╣┤7µ£ł’╝īõĖÄĶĄøĶ»║ĶÅ▓ĶŠŠµłÉÕżÜķĪ╣Ķ«ĪÕłÆńÜ䵳śńĢźÕÉłõĮ£’╝īĶÄĘÕŠŚ1.5õ║┐ńŠÄÕģāńÜäķóäõ╗śµ¼Š’╝īÕ╣ČÕÅ»ĶāĮĶÄĘÕŠŚĶČģĶ┐ć20õ║┐ńŠÄÕģāńÜäµĮ£Õ£©Õ╝ĆÕÅæŃĆüńøæń«ĪÕÆīķöĆÕö«ķćīń©ŗńóæ’╝īõ╗źÕÅŖÕÅ»Ķ¦éńÜäńē╣Ķ«ĖµØāõĮ┐ńö©Ķ┤╣ŃĆé
’╝łõ║ī’╝ē┬Ā ┬Ā ÕøĮÕåģ
Õ╝ƵŗōĶŹ»õĖÜ’╝ł9939.HK’╝ēµłÉń½ŗõ║Ä2009Õ╣┤’╝īõ╗źķøäµ┐Ćń┤ĀÕÅŚõĮō(AR)ÕÆīĶé┐ńśżńøĖÕģ│ń¢ŠńŚģõĖ║µĀĖÕ┐ā’╝īńĀöÕÅæÕżÜķĆÜķüōõ║¦Õōüń╗äÕÉł’╝īõ║¦ÕōüĶ”åńø¢Õģ©ńÉāķ½śÕÅæńŚģńÄćńÖīńŚćŃĆüµ¢░ÕåĀŃĆüĶä▒ÕÅæÕÆīńŚżń¢«ńŁēŃĆé2021Õ╣┤2µ£ł’╝īÕ╝ƵŗōĶŹ»õĖÜÕ«ŻÕĖāÕģČĶć¬õĖ╗ńĀöÕÅæńÜäÕģ©ńÉāķ”¢õĖ¬Õ¤║õ║ÄPROTACµŖƵ£»ńÜäÕż¢ńö©ARķÖŹĶ¦ŻÕēé’╝łGT20029’╝ēńÜäINDńö│Ķ»ĘÕĘ▓ĶÄĘÕøĮÕ«ČĶŹ»ÕōüńøæńØŻń«ĪńÉåÕ▒ĆÕÅŚńÉåŃĆéĶ»źÕģ¼ÕÅĖÕ░åķĆÜĶ┐ćÕ╝ĆÕÅæķØ×ÕÅŻµ£ŹńÜäPROTACĶŹ»ńē®µØźķ¬īĶ»üĶ┐ÖõĖĆÕ╝ĆÕłøµĆ¦ńÜäµŖƵ£»µś»ÕÉ”ÕÅ»õ╗źµłÉĶŹ»’╝īÕÉīµŚČõ╣¤µŁŻÕ£©ńĀöÕÅæÕÅŻµ£ŹńÜäķØČÕÉæARńÜäPROTAC’╝īķĆéÕ║öĶ»üõĖ║ķøäµ┐Ćń┤ĀµĆ¦Ķä▒ÕÅæÕÆīńŚżń¢«ńŁēŃĆé
µĄĘµĆØń¦æ’╝ł002653’╝ēµłÉń½ŗõ║Ä2000Õ╣┤’╝īÕģ¼ÕÅĖÕģʵ£ēµ»öĶŠāÕ«īÕ¢äńÜäPROTACÕ╣│ÕÅ░’╝īńö©õ╗źÕ╝ĆÕÅæķÆłÕ»╣Ķé┐ńśżÕÆīĶć¬Ķ║½ÕģŹń¢½µĆ¦ń¢ŠńŚģńÜäķ½śķĆēµŗ®µĆ¦õĖöÕÅŻµ£Źµ£ēµĢłńÜäĶøŗńÖĮķÖŹĶ¦ŻĶŹ»ńē®ŃĆéńø«ÕēŹPROTACńÜäķĪ╣ńø«µ£ēĶČģĶ┐ć20õĖ¬’╝īÕĖāÕ▒ĆńÜäķØČńé╣µ£ēBTKŃĆüIRAK4ŃĆüBRD4ŃĆüARŃĆüEGFRŃĆüKRASŃĆüALKŃĆüCDK4/6ŃĆüEZH2ŃĆüPARPŃĆüBcr/AblŃĆüSTAT3ńŁēŃĆé
2017Õ╣┤’╝īÕÆīÕŠäÕī╗ĶŹ»Õ£©õĖŖµĄĘń¦æµŖĆÕż¦ÕŁ”ÕåģµłÉń½ŗ’╝īÕģ¼ÕÅĖĶ»×ńö¤õ╣ŗÕłØÕ░▒ĶÄĘÕŠŚõ║åµØźĶć¬ĶüöÕÆīĶĄäµ£¼ŃĆüĶŹ»µśÄÕ║ĘÕŠĘŃĆüÕåŹķ╝ÄÕī╗ĶŹ»ŃĆüÕŹāķ¬źĶĄäµ£¼ńÜäµŖĢĶĄä’╝ī2018Õ╣┤ÕÅłÕ«ŻÕĖāµÉ║µēŗõĖŖµĄĘń¦æµŖĆÕż¦ÕŁ”ÕÉłõĮ£Õ╝ĆÕÅæŌĆ£ķģŹõĮōÕ»╝ÕÉæĶøŗńÖĮĶ┤©ķÖŹĶ¦ŻĶŹ»ńē®ŌĆØ’╝īÕ╣ČĶŠŠµłÉõ║åķĆŠõĖĆõ║┐ńŠÄķćæńÜ䵳śńĢźÕÉłõĮ£ÕŹÅĶ««ŃĆéÕģ¼ÕÅĖķĆÜĶ┐ćõĖÄõĖŖµĄĘń¦æµŖĆÕż¦ÕŁ”ńÜäķĪ╣ńø«ÕÉłõĮ£ĶĄĘµŁź’╝īÕ£©µē®Õż¦õ║¦Õōüń«Īń║┐ńÜäÕÉīµŚČµŁŻÕ£©ÕŖ¬ÕŖøµēōķĆĀĶć¬ÕĘ▒ńŗ¼ńē╣ńÜäµ¢░ĶŹ»ńĀöÕÅæÕ╣│ÕÅ░ŃĆé
ÕÆīÕŠäÕī╗ĶŹ»ńÜäPROTACÕ╣│ÕÅ░ĶĄĘµŁźĶŠāµŚ®’╝īń¼¼õĖĆõĖ¬PROTACÕÉłõĮ£ķĪ╣ńø«ÕĘ▓ń╗ÅńĀöÕÅæĶČģĶ┐ć3Õ╣┤µŚČķŚ┤’╝īńø«ÕēŹÕĘ▓ńŁøķĆēÕć║õ║åÕģʵ£ēķ½śµłÉĶŹ»µĆ¦ńÜäÕłåÕŁÉń│╗ÕłŚ’╝īĶ┐øÕ▒ĢĶŠāÕ┐½ńÜäÕłåÕŁÉÕŹ│Õ░åÕ╝ĆÕ▒ĢIND-EnablingŃĆéĶ»źÕłåÕŁÉń│╗ÕłŚÕģʵ£ēķ½śÕÅŻµ£Źńö¤ńē®Õł®ńö©Õ║”’╝īÕ╣ČÕ£©ÕŖ©ńē®õĮōÕåģĶŹ»µĢłµśŠĶæŚ’╝īÕ£©Õģŗµ£ŹķØ×Õ░Åń╗åĶā×Ķé║ńÖīÕ░ÅÕłåÕŁÉĶŹ»ńē®ĶĆÉĶŹ»ńŁēķóåÕ¤¤Õżäõ║ÄķóåÕģłÕ£░õĮŹŃĆé
ńÅāĶ»║ńö¤ńē®µłÉń½ŗõ║Ä2018Õ╣┤’╝īµś»õĖĆÕ«ČõĖōµ│©õ║ÄÕ░ÅÕłåÕŁÉÕłøµ¢░ĶŹ»ńĀöÕÅæńÜäńö¤ńē®Õī╗ĶŹ»õ╝üõĖÜŃĆéÕģ¼ÕÅĖõĖōµ│©õ║ÄÕ╝ĆÕÅæÕłåÕŁÉõ╝┤õŠŻõ╗ŗÕ»╝ńÜäĶøŗńÖĮĶ┤©ķÖŹĶ¦Ż’╝łChaperone-mediated Protein Degradation/Degrader, CHAMPTM’╝ēµŖƵ£»Õ╣│ÕÅ░’╝īńø«ÕēŹÕĘ▓µ£ēÕżÜõĖ¬Õģ©ńÉāÕłøµ¢░ńÜäµ¢░õĖĆń▒╗Õ░ÅÕłåÕŁÉµŖŚĶé┐ńśżÕī¢ÕÉłńē®ÕŹ│Õ░åĶ┐øÕģźõĖ┤Õ║Ŗµł¢Õżäõ║ÄõĖ┤Õ║ŖÕēŹÕ╝ĆÕÅæķśČµ«ĄŃĆé
õĮ£õĖ║ķØ®ÕæĮµĆ¦µŖƵ£»’╝īPROTACńÜäÕÅæÕ▒Ģń╗ÅÕÄåõ║å20Õ╣┤’╝īÕ░żÕģČÕ£©Ķ┐ćÕÄ╗ńÜä5Õ╣┤ÕÅæÕ▒ĢĶ┐ģńīø’╝īõ┐©ńäČÕĘ▓ń╗ŵłÉõĖ║µ¢░ĶŹ»ńĀöÕÅæńÜäµ¢░ķŻÄÕÅŻŃĆéÕł®ńö©Ķ»źµŖƵ£»ÕÅ»ÕĖāÕ▒ĆńÜäķØČńé╣Õ╣┐ķśö’╝īÕĖéÕ£║ÕĘ©Õż¦’╝īµ£¬µØźÕÅ»µ£¤ŃĆéõĮåµś»PROTACÕłåÕŁÉķćÅÕż¦ŃĆüńö¤ńē®Õł®ńö©Õ║”ÕĘ«ŃĆüµłÉĶŹ»Õø░ķÜŠńÜäńŚøńé╣õ╣¤ÕŠłµśÄµśŠ’╝īµ£¤ÕŠģĶ»źµŖƵ£»ńÜäõĖŹµ¢ŁĶ┐øµŁźÕÆīÕ«īÕ¢äŃĆéńøĖõ┐ĪķÜÅńØƵłÉĶŹ»µĆ¦ÕĘ«ńÜäķÜŠķóśĶó½µö╗Õģŗ’╝īPROTACÕÅ»õ╗źµłÉõĖ║ÕāÅÕ░ÅÕłåÕŁÉµŖæÕłČÕēéŃĆüÕŹĢµŖŚÕÆīÕģŹń¢½µ▓╗ń¢ŚńŁēõĖƵĀʵłÉÕŖ¤ńÜäń¢Śµ│ĢŃĆéÕĮōńäČ’╝īõ╣¤µ£¤ÕŠģń£ŗĶ¦üµø┤ÕżÜPROTACĶŹ»ńē®õĖŖÕĖé’╝īÕ╗ČķĢ┐µéŻĶĆģńö¤ÕæĮÕ╣ȵÅÉķ½śµéŻĶĆģńÜäńö¤ÕŁśĶ┤©ķćÅŃĆé
ÕģŹĶ┤ŻÕŻ░µśÄ
ńøĖÕģ│ÕåģÕ«╣Õ¤║õ║ÄÕĘ▓Õģ¼Õ╝ĆńÜäĶĄäµ¢Öµł¢õ┐Īµü»µÆ░ÕåÖ’╝īõĮåµ£¼Õģ¼ÕÅĖõĖŹõ┐ØĶ»üĶ»źńŁēõ┐Īµü»ÕÅŖĶĄäµ¢ÖńÜäÕ«īµĢ┤µĆ¦ŃĆüÕćåńĪ«µĆ¦’╝īµēĆÕɽõ┐Īµü»ÕÅŖĶĄäµ¢Öõ┐صīüÕ£©µ£Ćµ¢░ńŖȵĆüŃĆéÕÉīµŚČ’╝īµ£¼Õģ¼ÕÅĖµ£ēµØāÕ»╣µ£¼µŖźÕæŖµēĆÕɽõ┐Īµü»Õ£©õĖŹÕÅæÕć║ķĆÜń¤źńÜäµāģÕĮóõĖŗÕüÜÕć║õ┐«µö╣’╝īķśģĶ»╗ĶĆģÕ║öÕĮōĶć¬ĶĪīÕģ│µ│©ńøĖÕ║öńÜäµø┤µ¢░µł¢õ┐«µö╣ŃĆé
Õ£©õ╗╗õĮĢµāģÕåĄõĖŗ’╝īµ£¼ń»ćµ¢ćń½ĀõĖŁńÜäõ┐Īµü»µł¢µēĆĶĪ©Ķ┐░ńÜäµäÅĶ¦üÕØćõĖŹµ×䵳ÉÕ»╣õ╗╗õĮĢõ║║ńÜäµŖĢĶĄäÕ╗║Ķ««’╝īµŚĀĶ«║µś»ÕÉ”ÕĘ▓ń╗ŵśÄńż║µł¢µÜŚńż║’╝īµ£¼µŖźÕæŖõĖŹĶāĮõĮ£õĖ║ķüōõ╣ēńÜäŃĆüĶ┤Żõ╗╗ńÜäÕÆīµ│ĢÕŠŗńÜäõŠØµŹ«µł¢ĶĆģÕćŁĶ»üŃĆéÕ£©õ╗╗õĮĢµāģÕåĄõĖŗ’╝īµ£¼Õģ¼ÕÅĖõ║”õĖŹÕ»╣õ╗╗õĮĢõ║║ÕøĀõĮ┐ńö©µ£¼µ¢ćń½ĀõĖŁńÜäõ╗╗õĮĢÕåģÕ«╣µēĆÕ╝ĢĶć┤ńÜäõ╗╗õĮĢµŹ¤Õż▒Ķ┤¤õ╗╗õĮĢĶ┤Żõ╗╗ŃĆéµ£¼µ¢ćń½Āõ╗ģõĖ║µ£¼Õģ¼ÕÅĖµēƵ£ē’╝īµ£¬ń╗Åõ║ŗÕģłõ╣”ķØóĶ«ĖÕÅ»’╝īõ╗╗õĮĢµ£║µ×äÕÆīõĖ¬õ║║õĖŹÕŠŚõ╗źõ╗╗õĮĢÕĮóÕ╝Åń┐╗ńēłŃĆüÕżŹÕłČŃĆüÕÅæĶĪ©ŃĆüĶĮ¼ÕÅæµł¢Õ╝Ģńö©µ£¼µŖźÕæŖńÜäõ╗╗õĮĢķā©ÕłåŃĆé




{kind=link}